Chapter 7 Human PBMCs (10X Genomics)
7.1 Introduction
This performs an analysis of the public PBMC ID dataset generated by 10X Genomics (Zheng et al. 2017), starting from the filtered count matrix.
7.3 Quality control
Cell calling implicitly serves as a QC step to remove libraries with low total counts and number of detected genes. Thus, we will only filter on the mitochondrial proportion.
library(scater)
stats <- high.mito <- list()
for (n in names(all.sce)) {
current <- all.sce[[n]]
is.mito <- grep("MT", rowData(current)$Symbol_TENx)
stats[[n]] <- perCellQCMetrics(current, subsets=list(Mito=is.mito))
high.mito[[n]] <- isOutlier(stats[[n]]$subsets_Mito_percent, type="higher")
all.sce[[n]] <- current[,!high.mito[[n]]]
}qcplots <- list()
for (n in names(all.sce)) {
current <- unfiltered[[n]]
colData(current) <- cbind(colData(current), stats[[n]])
current$discard <- high.mito[[n]]
qcplots[[n]] <- plotColData(current, x="sum", y="subsets_Mito_percent",
colour_by="discard") + scale_x_log10()
}
do.call(gridExtra::grid.arrange, c(qcplots, ncol=3))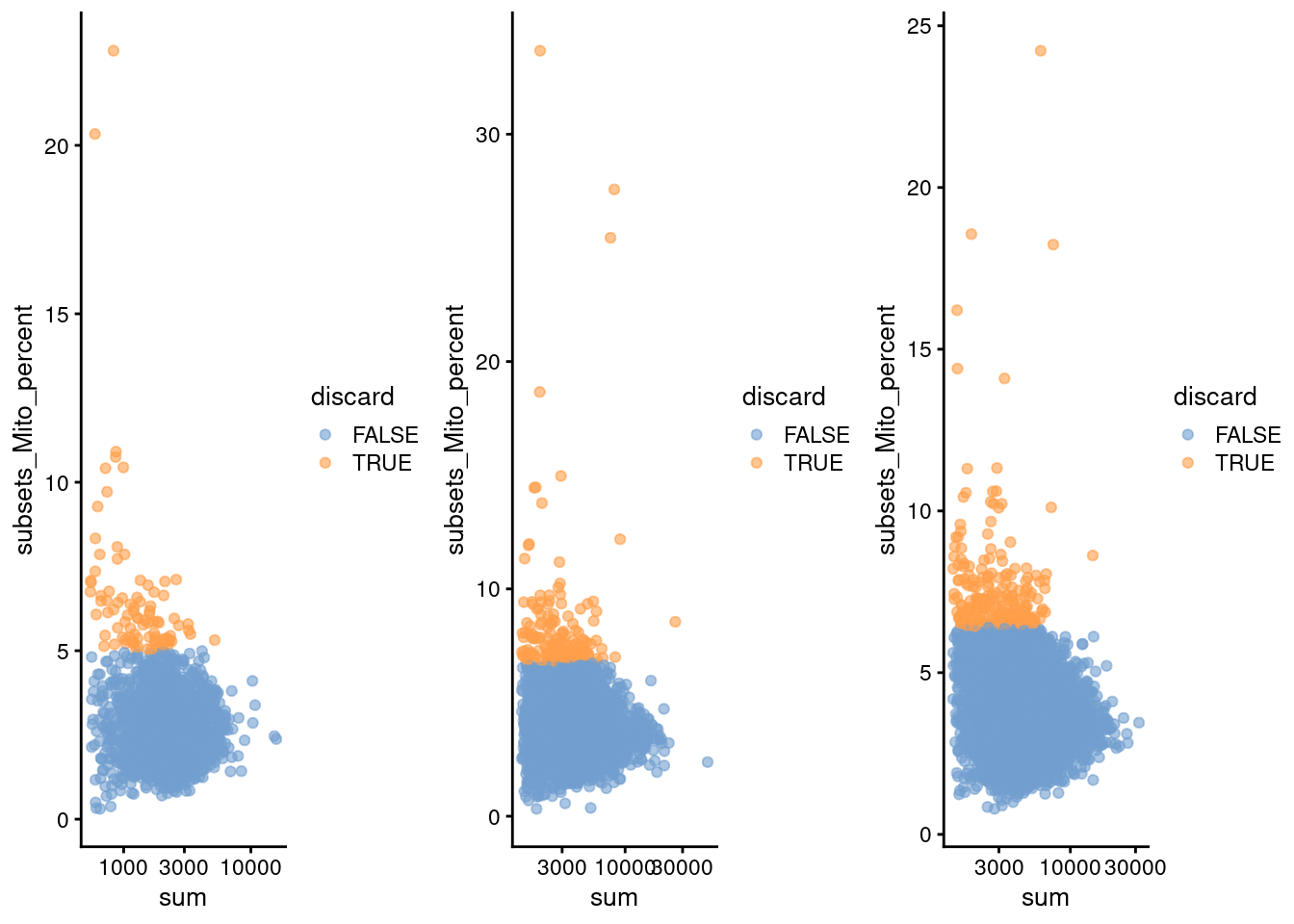
Figure 7.1: Percentage of mitochondrial reads in each cell in each of the 10X PBMC datasets, compared to the total count. Each point represents a cell and is colored according to whether that cell was discarded.
## $pbmc3k
## Mode FALSE TRUE
## logical 2609 91
##
## $pbmc4k
## Mode FALSE TRUE
## logical 4182 158
##
## $pbmc8k
## Mode FALSE TRUE
## logical 8157 2247.4 Normalization
We perform library size normalization, simply for convenience when dealing with file-backed matrices.
## $pbmc3k
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.234 0.748 0.926 1.000 1.157 6.604
##
## $pbmc4k
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.315 0.711 0.890 1.000 1.127 11.027
##
## $pbmc8k
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.296 0.704 0.877 1.000 1.118 6.7947.5 Variance modelling
library(scran)
all.dec <- lapply(all.sce, modelGeneVar)
all.hvgs <- lapply(all.dec, getTopHVGs, prop=0.1)par(mfrow=c(1,3))
for (n in names(all.dec)) {
curdec <- all.dec[[n]]
plot(curdec$mean, curdec$total, pch=16, cex=0.5, main=n,
xlab="Mean of log-expression", ylab="Variance of log-expression")
curfit <- metadata(curdec)
curve(curfit$trend(x), col='dodgerblue', add=TRUE, lwd=2)
}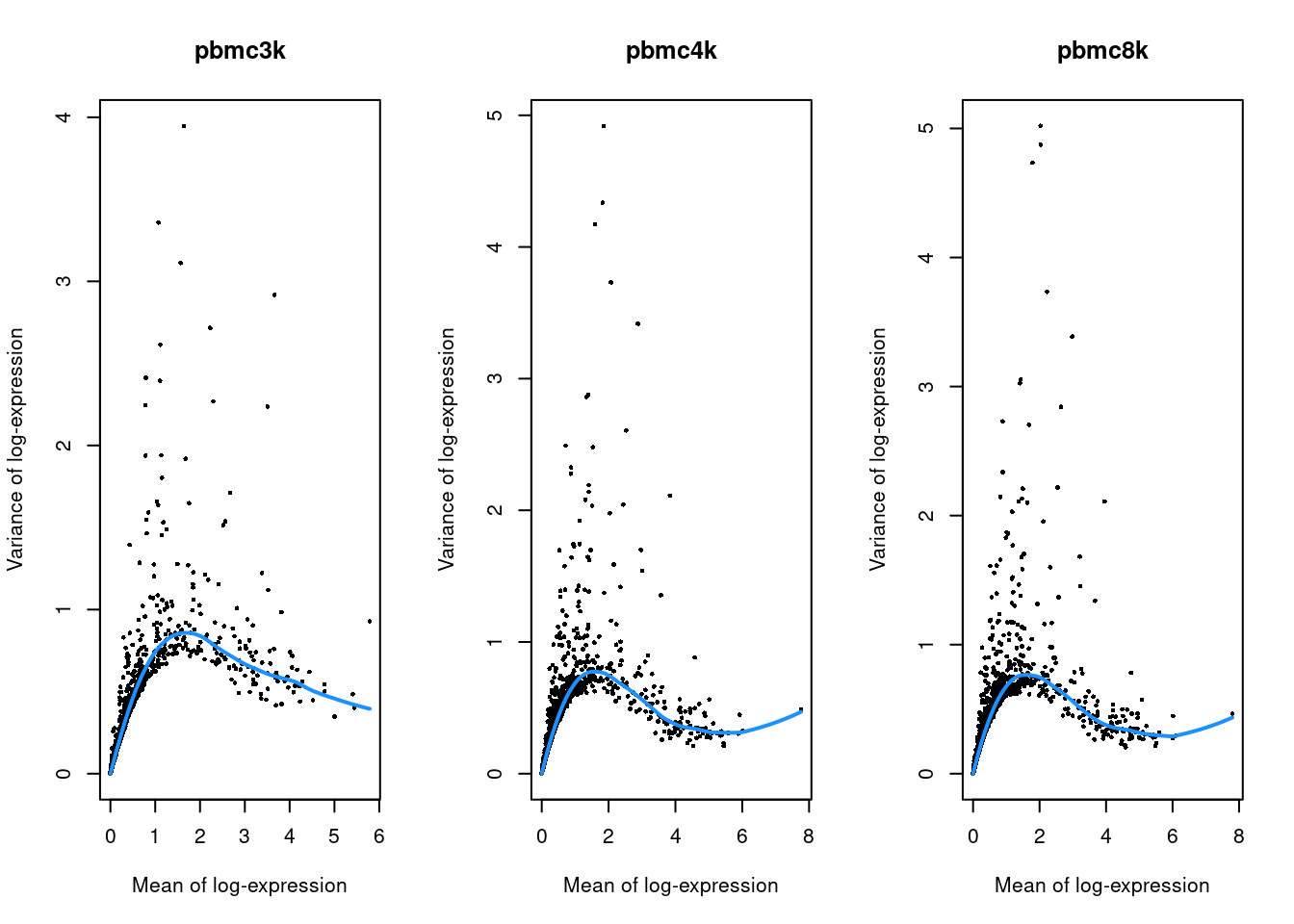
Figure 7.2: Per-gene variance as a function of the mean for the log-expression values in each PBMC dataset. Each point represents a gene (black) with the mean-variance trend (blue) fitted to the variances.
7.6 Dimensionality reduction
For various reasons, we will first analyze each PBMC dataset separately rather than merging them together. We use randomized SVD, which is more efficient for file-backed matrices.
library(BiocSingular)
set.seed(10000)
all.sce <- mapply(FUN=runPCA, x=all.sce, subset_row=all.hvgs,
MoreArgs=list(ncomponents=25, BSPARAM=RandomParam()),
SIMPLIFY=FALSE)
set.seed(100000)
all.sce <- lapply(all.sce, runTSNE, dimred="PCA")
set.seed(1000000)
all.sce <- lapply(all.sce, runUMAP, dimred="PCA")7.7 Clustering
for (n in names(all.sce)) {
g <- buildSNNGraph(all.sce[[n]], k=10, use.dimred='PCA')
clust <- igraph::cluster_walktrap(g)$membership
colLabels(all.sce[[n]]) <- factor(clust)
}## $pbmc3k
##
## 1 2 3 4 5 6 7 8 9 10
## 475 636 153 476 164 31 159 164 340 11
##
## $pbmc4k
##
## 1 2 3 4 5 6 7 8 9 10 11 12
## 127 594 518 775 211 394 187 993 55 201 91 36
##
## $pbmc8k
##
## 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15
## 292 1603 388 94 738 1035 1049 156 203 153 2098 261 64 14 9all.tsne <- list()
for (n in names(all.sce)) {
all.tsne[[n]] <- plotTSNE(all.sce[[n]], colour_by="label") + ggtitle(n)
}
do.call(gridExtra::grid.arrange, c(all.tsne, list(ncol=2)))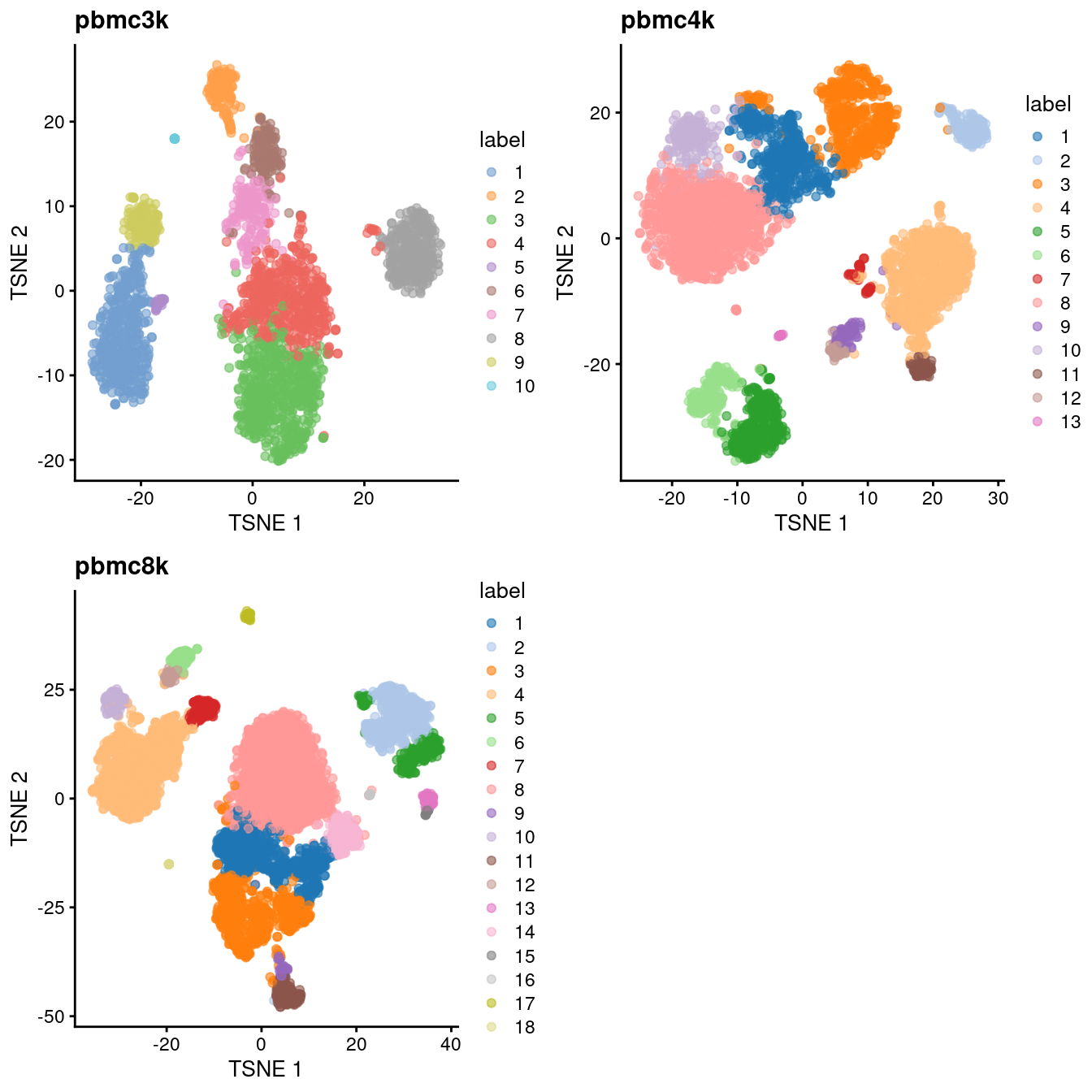
Figure 7.3: Obligatory \(t\)-SNE plots of each PBMC dataset, where each point represents a cell in the corresponding dataset and is colored according to the assigned cluster.
7.8 Data integration
With the per-dataset analyses out of the way, we will now repeat the analysis after merging together the three batches.
# Intersecting the common genes.
universe <- Reduce(intersect, lapply(all.sce, rownames))
all.sce2 <- lapply(all.sce, "[", i=universe,)
all.dec2 <- lapply(all.dec, "[", i=universe,)
# Renormalizing to adjust for differences in depth.
library(batchelor)
normed.sce <- do.call(multiBatchNorm, all.sce2)
# Identifying a set of HVGs using stats from all batches.
combined.dec <- do.call(combineVar, all.dec2)
combined.hvg <- getTopHVGs(combined.dec, n=5000)
set.seed(1000101)
merged.pbmc <- do.call(fastMNN, c(normed.sce,
list(subset.row=combined.hvg, BSPARAM=RandomParam())))We use the percentage of lost variance as a diagnostic measure.
## pbmc3k pbmc4k pbmc8k
## [1,] 7.044e-03 3.129e-03 0.000000
## [2,] 6.876e-05 4.912e-05 0.003008We proceed to clustering:
g <- buildSNNGraph(merged.pbmc, use.dimred="corrected")
colLabels(merged.pbmc) <- factor(igraph::cluster_louvain(g)$membership)
table(colLabels(merged.pbmc), merged.pbmc$batch)##
## pbmc3k pbmc4k pbmc8k
## 1 535 426 830
## 2 331 588 1126
## 3 182 122 217
## 4 150 179 292
## 5 170 345 573
## 6 292 538 1020
## 7 342 630 1236
## 8 437 749 1538
## 9 9 18 95
## 10 97 365 782
## 11 34 120 201
## 12 11 54 159
## 13 11 3 9
## 14 4 36 64
## 15 4 9 15And visualization:
set.seed(10101010)
merged.pbmc <- runTSNE(merged.pbmc, dimred="corrected")
gridExtra::grid.arrange(
plotTSNE(merged.pbmc, colour_by="label", text_by="label", text_colour="red"),
plotTSNE(merged.pbmc, colour_by="batch")
)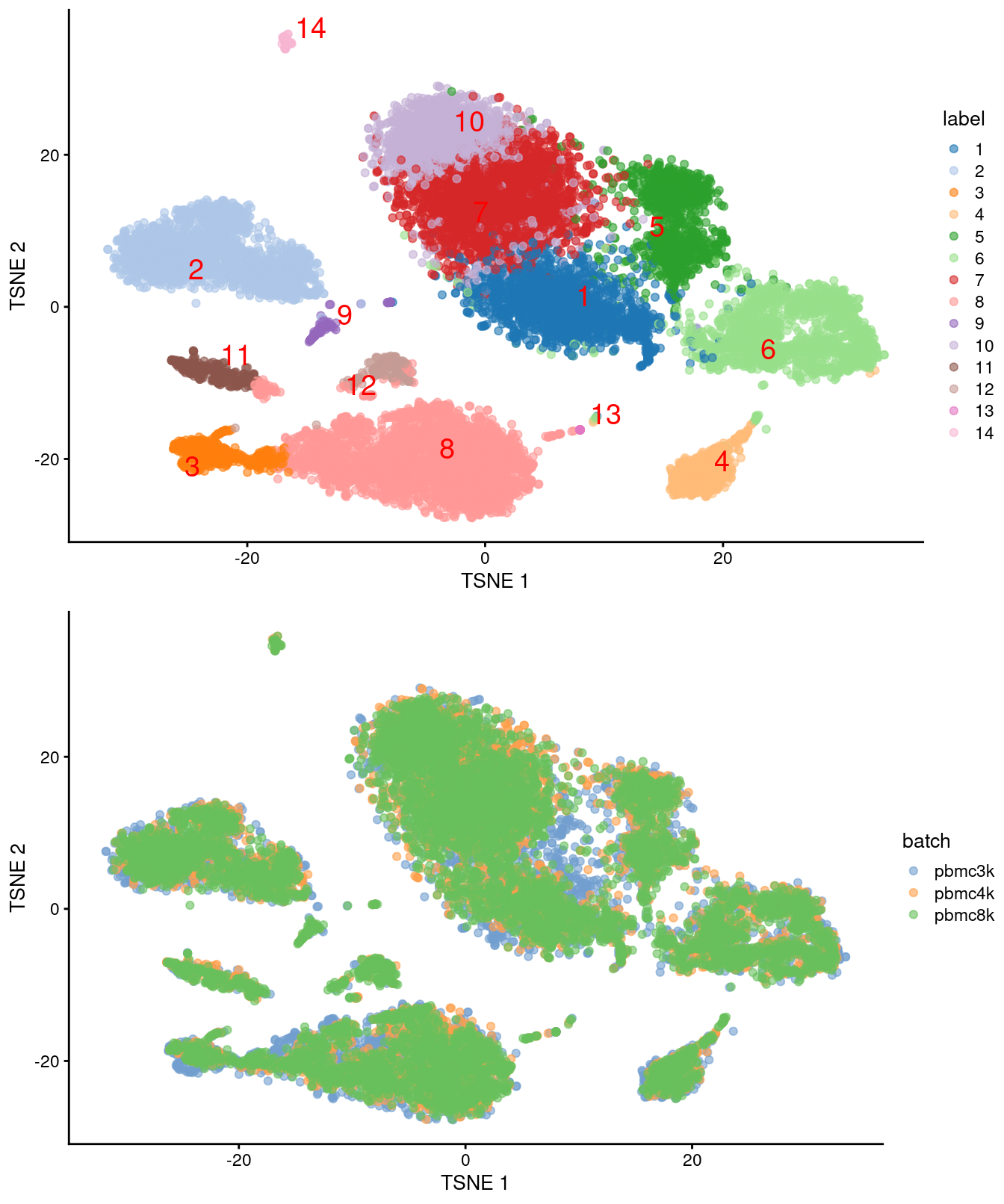
Figure 7.4: Obligatory \(t\)-SNE plots for the merged PBMC datasets, where each point represents a cell and is colored by cluster (top) or batch (bottom).
Session Info
R version 4.6.0 RC (2026-04-17 r89917)
Platform: x86_64-pc-linux-gnu
Running under: Ubuntu 24.04.4 LTS
Matrix products: default
BLAS: /home/biocbuild/bbs-3.23-bioc/R/lib/libRblas.so
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.12.0 LAPACK version 3.12.0
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_GB LC_COLLATE=C
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
time zone: America/New_York
tzcode source: system (glibc)
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] batchelor_1.28.0 BiocSingular_1.28.0
[3] scran_1.40.0 scater_1.40.0
[5] ggplot2_4.0.3 scuttle_1.22.0
[7] TENxPBMCData_1.29.0 HDF5Array_1.40.0
[9] h5mread_1.4.0 rhdf5_2.56.0
[11] DelayedArray_0.38.0 SparseArray_1.12.0
[13] S4Arrays_1.12.0 abind_1.4-8
[15] Matrix_1.7-5 SingleCellExperiment_1.34.0
[17] SummarizedExperiment_1.42.0 Biobase_2.72.0
[19] GenomicRanges_1.64.0 Seqinfo_1.2.0
[21] IRanges_2.46.0 S4Vectors_0.50.0
[23] BiocGenerics_0.58.0 generics_0.1.4
[25] MatrixGenerics_1.24.0 matrixStats_1.5.0
[27] BiocStyle_2.40.0 rebook_1.22.0
loaded via a namespace (and not attached):
[1] RColorBrewer_1.1-3 jsonlite_2.0.0
[3] CodeDepends_0.6.7 magrittr_2.0.5
[5] ggbeeswarm_0.7.3 farver_2.1.2
[7] rmarkdown_2.31 vctrs_0.7.3
[9] DelayedMatrixStats_1.34.0 memoise_2.0.1
[11] htmltools_0.5.9 AnnotationHub_4.2.0
[13] curl_7.1.0 BiocNeighbors_2.6.0
[15] Rhdf5lib_2.0.0 sass_0.4.10
[17] bslib_0.10.0 httr2_1.2.2
[19] cachem_1.1.0 ResidualMatrix_1.22.0
[21] igraph_2.3.0 lifecycle_1.0.5
[23] pkgconfig_2.0.3 rsvd_1.0.5
[25] R6_2.6.1 fastmap_1.2.0
[27] digest_0.6.39 AnnotationDbi_1.74.0
[29] dqrng_0.4.1 RSpectra_0.16-2
[31] irlba_2.3.7 ExperimentHub_3.2.0
[33] RSQLite_2.4.6 beachmat_2.28.0
[35] filelock_1.0.3 labeling_0.4.3
[37] httr_1.4.8 compiler_4.6.0
[39] bit64_4.8.0 withr_3.0.2
[41] S7_0.2.2 BiocParallel_1.46.0
[43] viridis_0.6.5 DBI_1.3.0
[45] rappdirs_0.3.4 bluster_1.22.0
[47] tools_4.6.0 vipor_0.4.7
[49] otel_0.2.0 beeswarm_0.4.0
[51] glue_1.8.1 rhdf5filters_1.24.0
[53] grid_4.6.0 Rtsne_0.17
[55] cluster_2.1.8.2 gtable_0.3.6
[57] ScaledMatrix_1.20.0 metapod_1.20.0
[59] XVector_0.52.0 RcppAnnoy_0.0.23
[61] ggrepel_0.9.8 BiocVersion_3.23.1
[63] pillar_1.11.1 limma_3.68.0
[65] dplyr_1.2.1 BiocFileCache_3.2.0
[67] lattice_0.22-9 FNN_1.1.4.1
[69] bit_4.6.0 tidyselect_1.2.1
[71] locfit_1.5-9.12 Biostrings_2.80.0
[73] knitr_1.51 gridExtra_2.3
[75] bookdown_0.46 edgeR_4.10.0
[77] xfun_0.57 statmod_1.5.1
[79] yaml_2.3.12 evaluate_1.0.5
[81] codetools_0.2-20 tibble_3.3.1
[83] BiocManager_1.30.27 graph_1.90.0
[85] cli_3.6.6 uwot_0.2.4
[87] jquerylib_0.1.4 dichromat_2.0-0.1
[89] Rcpp_1.1.1-1.1 dir.expiry_1.20.0
[91] dbplyr_2.5.2 png_0.1-9
[93] XML_3.99-0.23 parallel_4.6.0
[95] blob_1.3.0 sparseMatrixStats_1.24.0
[97] beachmat.hdf5_1.10.0 viridisLite_0.4.3
[99] scales_1.4.0 purrr_1.2.2
[101] crayon_1.5.3 rlang_1.2.0
[103] cowplot_1.2.0 KEGGREST_1.52.0 References
Zheng, G. X., J. M. Terry, P. Belgrader, P. Ryvkin, Z. W. Bent, R. Wilson, S. B. Ziraldo, et al. 2017. “Massively parallel digital transcriptional profiling of single cells.” Nat Commun 8 (January): 14049.