Chapter 8 Annotation and visualization
8.1 Adding gene-based annotation
Annotation can be added to a given set of regions using the detailRanges() function.
This will identify overlaps between the regions and annotated genomic features such as exons, introns and promoters.
Here, the promoter region of each gene is defined as some interval 3 kbp up- and 1 kbp downstream of the TSS for that gene.
Any exonic features within dist on the left or right side of each supplied region will also be reported.
#--- loading-files ---#
library(chipseqDBData)
tf.data <- NFYAData()
tf.data
bam.files <- head(tf.data$Path, -1) # skip the input.
bam.files
#--- counting-windows ---#
library(csaw)
frag.len <- 110
win.width <- 10
param <- readParam(minq=20)
data <- windowCounts(bam.files, ext=frag.len, width=win.width, param=param)
#--- filtering ---#
binned <- windowCounts(bam.files, bin=10000, param=param)
fstats <- filterWindowsGlobal(data, binned)
filtered.data <- data[fstats$filter > log2(5),]
#--- normalization ---#
filtered.data <- normFactors(binned, se.out=filtered.data)
#--- modelling ---#
cell.type <- sub("NF-YA ([^ ]+) .*", "\\1", head(tf.data$Description, -1))
design <- model.matrix(~cell.type)
colnames(design) <- c("intercept", "cell.type")
library(edgeR)
y <- asDGEList(filtered.data)
y <- estimateDisp(y, design)
fit <- glmQLFit(y, design, robust=TRUE)
res <- glmQLFTest(fit, coef="cell.type")
rowData(filtered.data) <- cbind(rowData(filtered.data), res$table)
#--- merging ---#
merged <- mergeResults(filtered.data, tol=1000,
merge.args=list(max.width=5000))library(csaw)
library(org.Mm.eg.db)
library(TxDb.Mmusculus.UCSC.mm10.knownGene)
anno <- detailRanges(merged$regions, txdb=TxDb.Mmusculus.UCSC.mm10.knownGene,
orgdb=org.Mm.eg.db, promoter=c(3000, 1000), dist=5000)
head(anno$overlap)## [1] "" "" "Rrs1:+:P,Adhfe1:+:P"
## [4] "Ppp1r42:-:I" "" "Ncoa2:-:PI"## [1] "" "" "" "Ppp1r42:-:3948"
## [5] "" "Ncoa2:-:12"## [1] "" "Rrs1:+:3898"
## [3] "Rrs1:+:48,Adhfe1:+:2588" "Ppp1r42:-:1612"
## [5] "Ncoa2:-:4595" "Ncoa2:-:1278"Character vectors of compact string representations are provided to summarize the features overlapped by each supplied region.
Each pattern contains GENE|STRAND|TYPE to describe the strand and overlapped features of that gene.
Exons are labelled as E, promoters are P and introns are I.
For left and right, TYPE is replaced by DISTANCE.
This indicates the gap (in base pairs) between the supplied region and the closest non-overlapping exon of the annotated feature.
All of this annotation can be stored in the metadata of the GRanges object for later use.
merged$regions$overlap <- anno$overlap
merged$regions$left <- anno$left
merged$regions$right <- anno$rightWhile the string representation saves space in the output, it is not easy to work with.
If the annotation needs to manipulated directly, users can obtain it from the detailRanges() command by not specifying the regions of interest.
This can then be used for interactive manipulation, e.g., to identify all genes where the promoter contains DB sites.
anno.ranges <- detailRanges(txdb=TxDb.Mmusculus.UCSC.mm10.knownGene,
orgdb=org.Mm.eg.db)
anno.ranges## GRanges object with 505738 ranges and 2 metadata columns:
## seqnames ranges strand | symbol type
## <Rle> <IRanges> <Rle> | <character> <character>
## 100009600 chr9 21062393-21062717 - | Zglp1 E
## 100009600 chr9 21062400-21062717 - | Zglp1 E
## 100009600 chr9 21062894-21062987 - | Zglp1 E
## 100009600 chr9 21063314-21063396 - | Zglp1 E
## 100009600 chr9 21066024-21066377 - | Zglp1 E
## ... ... ... ... . ... ...
## 99982 chr4 136554325-136558324 - | Kdm1a P
## 99982 chr4 136560502-136564501 - | Kdm1a P
## 99982 chr4 136567608-136571607 - | Kdm1a P
## 99982 chr4 136576159-136580158 - | Kdm1a P
## 99982 chr4 136550540-136602723 - | Kdm1a G
## -------
## seqinfo: 66 sequences (1 circular) from mm10 genome8.2 Checking bimodality for TF studies
For TF experiments, a simple measure of strand bimodality can be reported as a diagnostic.
Given a set of regions, the checkBimodality() function will return the maximum bimodality score across all base positions in each region.
The bimodality score at each base position is defined as the minimum of the ratio of the number of forward- to reverse-stranded reads to the left of that position, and the ratio of the reverse- to forward-stranded reads to the right.
A high score is only possible if both ratios are large, i.e., strand bimodality is present.
# TODO: make this less weird.
spacing <- metadata(data)$spacing
expanded <- resize(merged$regions, fix="center",
width=width(merged$regions)+spacing)
sbm.score <- checkBimodality(bam.files, expanded, width=frag.len)
head(sbm.score)## [1] 1.456 2.263 1.364 5.333 1.933 3.405In the above code, all regions are expanded by spacing, i.e., 50 bp.
This ensures that the optimal bimodality score can be computed for the centre of the binding site, even if that position is not captured by a window.
The width argument specifies the span with which to count reads for the score calculation.
This should be set to the average fragment length.
If multiple bam.files are provided, they will be pooled during counting.
For typical TF binding sites, bimodality scores can be considered to be “high” if they are larger than 4. This allows users to distinguish between genuine binding sites and high-abundance artifacts such as repeats or read stacks. However, caution is still required as some high scores may be driven by the stochastic distribution of reads. Obviously, the concept of strand bimodality is less relevant for diffuse targets like histone marks.
8.3 Saving the results to file
It is a simple matter to save the results for later perusal, e.g., to a tab-separated file.
ofile <- gzfile("clusters.tsv.gz", open="w")
write.table(as.data.frame(merged), file=ofile,
row.names=FALSE, quote=FALSE, sep="\t")
close(ofile)Of course, other formats can be used depending on the purpose of the file. For example, significantly DB regions can be exported to BED files through the rtracklayer package for visual inspection with genomic browsers. A transformed FDR is used here for the score field.
is.sig <- merged$combined$FDR <= 0.05
test <- merged$regions[is.sig]
test$score <- -10*log10(merged$combined$FDR[is.sig])
names(test) <- paste0("region", 1:sum(is.sig))
library(rtracklayer)
export(test, "clusters.bed")
head(read.table("clusters.bed"))## V1 V2 V3 V4 V5 V6
## 1 chr1 7397900 7398110 region1 13.74 .
## 2 chr1 9541400 9541510 region2 15.60 .
## 3 chr1 13589950 13590010 region3 14.56 .
## 4 chr1 15805500 15805660 region4 22.60 .
## 5 chr1 23256050 23256210 region5 14.47 .
## 6 chr1 32172600 32172710 region6 13.82 .Alternatively, the GRanges object can be directly saved to file and reloaded later for direct manipulation in the R environment, e.g., to find overlaps with other regions of interest.
8.4 Simple visualization of genomic coverage
Visualization of the read depth around interesting features is often desired.
This is facilitated by the extractReads() function, which pulls out the reads from the BAM file.
The returned GRanges object can then be used to plot the sequencing coverage or any other statistic of interest.
Note that the extractReads() function also accepts a readParam object.
This ensures that the same reads used in the analysis will be pulled out during visualization.
cur.region <- GRanges("chr18", IRanges(77806807, 77807165))
extractReads(bam.files[[1]], cur.region, param=param)## GRanges object with 55 ranges and 0 metadata columns:
## seqnames ranges strand
## <Rle> <IRanges> <Rle>
## [1] chr18 77806886-77806922 +
## [2] chr18 77806887-77806923 +
## [3] chr18 77806887-77806923 +
## [4] chr18 77806887-77806923 +
## [5] chr18 77806890-77806926 +
## ... ... ... ...
## [51] chr18 77807063-77807095 -
## [52] chr18 77807068-77807104 -
## [53] chr18 77807082-77807119 -
## [54] chr18 77807084-77807120 -
## [55] chr18 77807087-77807123 -
## -------
## seqinfo: 1 sequence from an unspecified genomeHere, coverage is visualized as the number of reads covering each base pair in the interval of interest. Specifically, the reads-per-million is shown to allow comparisons between libraries of different size. The plots themselves are constructed using methods from the Gviz package. The blue and red tracks represent the coverage on the forward and reverse strands, respectively. Strong strand bimodality is consistent with a genuine TF binding site. For paired-end data, coverage can be similarly plotted for fragments, i.e., proper read pairs.
library(Gviz)
collected <- vector("list", length(bam.files))
for (i in seq_along(bam.files)) {
reads <- extractReads(bam.files[[i]], cur.region, param=param)
adj.total <- data$totals[i]/1e6
pcov <- as(coverage(reads[strand(reads)=="+"])/adj.total, "GRanges")
ncov <- as(coverage(reads[strand(reads)=="-"])/adj.total, "GRanges")
ptrack <- DataTrack(pcov, type="histogram", lwd=0, fill=rgb(0,0,1,.4),
ylim=c(0,1.1), name=tf.data$Name[i], col.axis="black",
col.title="black")
ntrack <- DataTrack(ncov, type="histogram", lwd=0, fill=rgb(1,0,0,.4),
ylim=c(0,1.1))
collected[[i]] <- OverlayTrack(trackList=list(ptrack,ntrack))
}
gax <- GenomeAxisTrack(col="black")
plotTracks(c(gax, collected), from=start(cur.region), to=end(cur.region))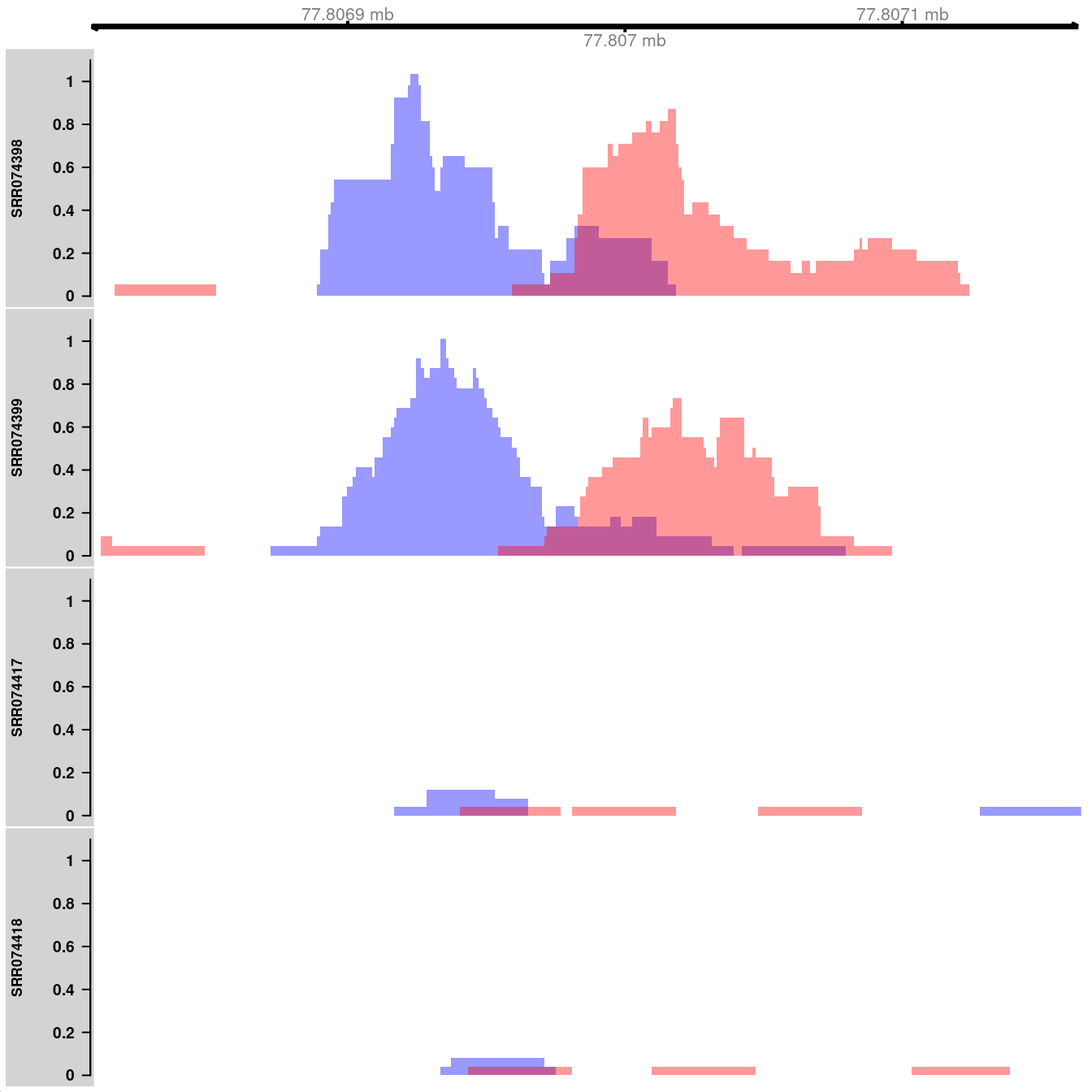
Session information
R version 4.2.1 (2022-06-23)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 20.04.4 LTS
Matrix products: default
BLAS: /home/biocbuild/bbs-3.15-bioc/R/lib/libRblas.so
LAPACK: /home/biocbuild/bbs-3.15-bioc/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_GB LC_COLLATE=C
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] grid stats4 stats graphics grDevices utils datasets
[8] methods base
other attached packages:
[1] Gviz_1.40.1
[2] rtracklayer_1.56.1
[3] TxDb.Mmusculus.UCSC.mm10.knownGene_3.10.0
[4] GenomicFeatures_1.48.3
[5] org.Mm.eg.db_3.15.0
[6] AnnotationDbi_1.58.0
[7] csaw_1.30.1
[8] SummarizedExperiment_1.26.1
[9] Biobase_2.56.0
[10] MatrixGenerics_1.8.1
[11] matrixStats_0.62.0
[12] GenomicRanges_1.48.0
[13] GenomeInfoDb_1.32.3
[14] IRanges_2.30.1
[15] S4Vectors_0.34.0
[16] BiocGenerics_0.42.0
[17] BiocStyle_2.24.0
[18] rebook_1.6.0
loaded via a namespace (and not attached):
[1] colorspace_2.0-3 deldir_1.0-6 rjson_0.2.21
[4] ellipsis_0.3.2 htmlTable_2.4.1 biovizBase_1.44.0
[7] XVector_0.36.0 base64enc_0.1-3 dichromat_2.0-0.1
[10] rstudioapi_0.14 bit64_4.0.5 fansi_1.0.3
[13] xml2_1.3.3 codetools_0.2-18 splines_4.2.1
[16] cachem_1.0.6 knitr_1.40 Formula_1.2-4
[19] jsonlite_1.8.0 Rsamtools_2.12.0 cluster_2.1.4
[22] dbplyr_2.2.1 png_0.1-7 graph_1.74.0
[25] BiocManager_1.30.18 compiler_4.2.1 httr_1.4.4
[28] backports_1.4.1 lazyeval_0.2.2 assertthat_0.2.1
[31] Matrix_1.4-1 fastmap_1.1.0 limma_3.52.2
[34] cli_3.3.0 htmltools_0.5.3 prettyunits_1.1.1
[37] tools_4.2.1 gtable_0.3.0 glue_1.6.2
[40] GenomeInfoDbData_1.2.8 dplyr_1.0.9 rappdirs_0.3.3
[43] Rcpp_1.0.9 jquerylib_0.1.4 vctrs_0.4.1
[46] Biostrings_2.64.1 xfun_0.32 stringr_1.4.1
[49] lifecycle_1.0.1 ensembldb_2.20.2 restfulr_0.0.15
[52] XML_3.99-0.10 edgeR_3.38.4 zlibbioc_1.42.0
[55] scales_1.2.1 BSgenome_1.64.0 VariantAnnotation_1.42.1
[58] ProtGenerics_1.28.0 hms_1.1.2 parallel_4.2.1
[61] AnnotationFilter_1.20.0 RColorBrewer_1.1-3 yaml_2.3.5
[64] curl_4.3.2 gridExtra_2.3 memoise_2.0.1
[67] ggplot2_3.3.6 sass_0.4.2 rpart_4.1.16
[70] biomaRt_2.52.0 latticeExtra_0.6-30 stringi_1.7.8
[73] RSQLite_2.2.16 highr_0.9 BiocIO_1.6.0
[76] checkmate_2.1.0 filelock_1.0.2 BiocParallel_1.30.3
[79] rlang_1.0.4 pkgconfig_2.0.3 bitops_1.0-7
[82] evaluate_0.16 lattice_0.20-45 purrr_0.3.4
[85] htmlwidgets_1.5.4 GenomicAlignments_1.32.1 CodeDepends_0.6.5
[88] bit_4.0.4 tidyselect_1.1.2 magrittr_2.0.3
[91] bookdown_0.28 R6_2.5.1 generics_0.1.3
[94] Hmisc_4.7-1 metapod_1.4.0 DelayedArray_0.22.0
[97] DBI_1.1.3 foreign_0.8-82 pillar_1.8.1
[100] nnet_7.3-17 survival_3.4-0 KEGGREST_1.36.3
[103] RCurl_1.98-1.8 tibble_3.1.8 dir.expiry_1.4.0
[106] crayon_1.5.1 interp_1.1-3 utf8_1.2.2
[109] BiocFileCache_2.4.0 rmarkdown_2.16 jpeg_0.1-9
[112] progress_1.2.2 locfit_1.5-9.6 data.table_1.14.2
[115] blob_1.2.3 digest_0.6.29 munsell_0.5.0
[118] bslib_0.4.0