Chapter 14 HCA human bone marrow (10X Genomics)
14.1 Introduction
Here, we use an example dataset from the Human Cell Atlas immune cell profiling project on bone marrow, which contains scRNA-seq data for 380,000 cells generated using the 10X Genomics technology. This is a fairly big dataset that represents a good use case for the techniques in Advanced Chapter 14.
14.2 Data loading
This dataset is loaded via the HCAData package, which provides a ready-to-use SingleCellExperiment object.
library(HCAData)
sce.bone <- HCAData('ica_bone_marrow', as.sparse=TRUE)
sce.bone$Donor <- sub("_.*", "", sce.bone$Barcode)We use symbols in place of IDs for easier interpretation later.
14.3 Quality control
Cell calling was not performed (see here) so we will perform QC using all metrics and block on the donor of origin during outlier detection. We perform the calculation across multiple cores to speed things up.
library(BiocParallel)
bpp <- MulticoreParam(8)
sce.bone <- unfiltered <- addPerCellQC(sce.bone, BPPARAM=bpp,
subsets=list(Mito=which(rowData(sce.bone)$Chr=="MT")))
qc <- quickPerCellQC(colData(sce.bone), batch=sce.bone$Donor,
sub.fields="subsets_Mito_percent")
sce.bone <- sce.bone[,!qc$discard]unfiltered$discard <- qc$discard
gridExtra::grid.arrange(
plotColData(unfiltered, x="Donor", y="sum", colour_by="discard") +
scale_y_log10() + ggtitle("Total count"),
plotColData(unfiltered, x="Donor", y="detected", colour_by="discard") +
scale_y_log10() + ggtitle("Detected features"),
plotColData(unfiltered, x="Donor", y="subsets_Mito_percent",
colour_by="discard") + ggtitle("Mito percent"),
ncol=2
)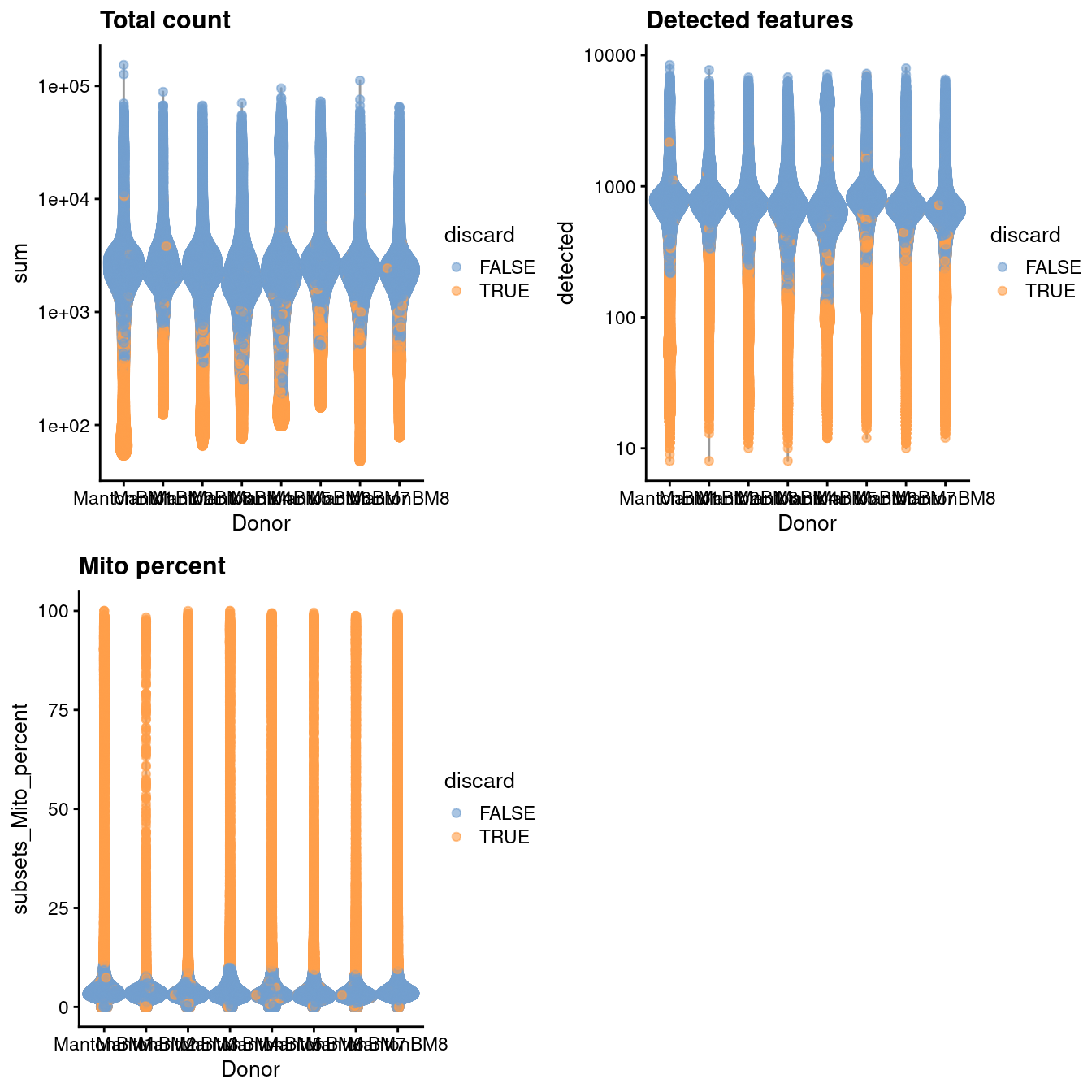
Figure 14.1: Distribution of QC metrics in the HCA bone marrow dataset. Each point represents a cell and is colored according to whether it was discarded.
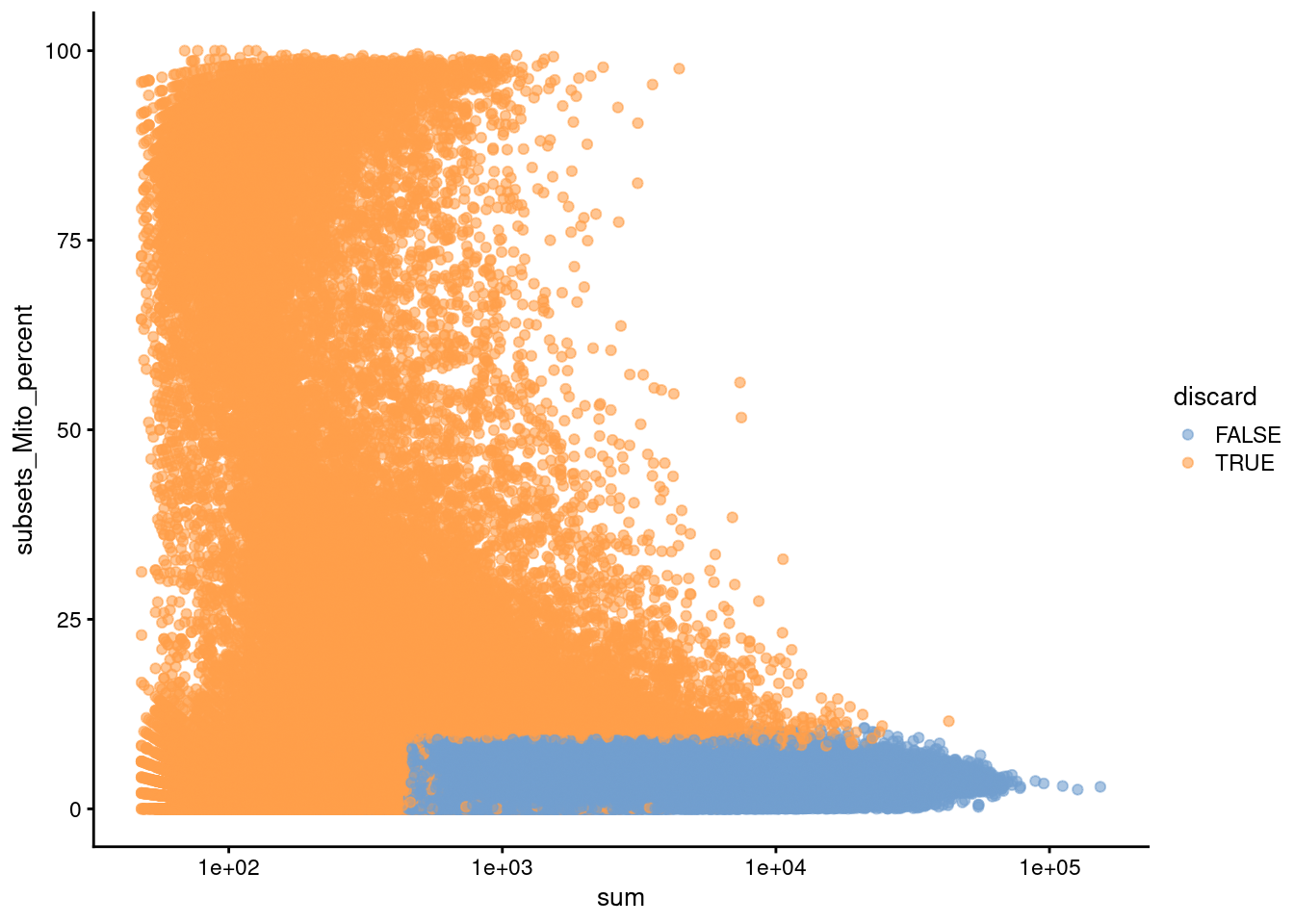
Figure 14.2: Percentage of mitochondrial reads in each cell in the HCA bone marrow dataset compared to its total count. Each point represents a cell and is colored according to whether that cell was discarded.
14.4 Normalization
For a minor speed-up, we use already-computed library sizes rather than re-computing them from the column sums.
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.05 0.47 0.65 1.00 0.89 42.3814.5 Variance modeling
We block on the donor of origin to mitigate batch effects during HVG selection. We select a larger number of HVGs to capture any batch-specific variation that might be present.
library(scran)
set.seed(1010010101)
dec.bone <- modelGeneVarByPoisson(sce.bone,
block=sce.bone$Donor, BPPARAM=bpp)
top.bone <- getTopHVGs(dec.bone, n=5000)par(mfrow=c(4,2))
blocked.stats <- dec.bone$per.block
for (i in colnames(blocked.stats)) {
current <- blocked.stats[[i]]
plot(current$mean, current$total, main=i, pch=16, cex=0.5,
xlab="Mean of log-expression", ylab="Variance of log-expression")
curfit <- metadata(current)
curve(curfit$trend(x), col='dodgerblue', add=TRUE, lwd=2)
}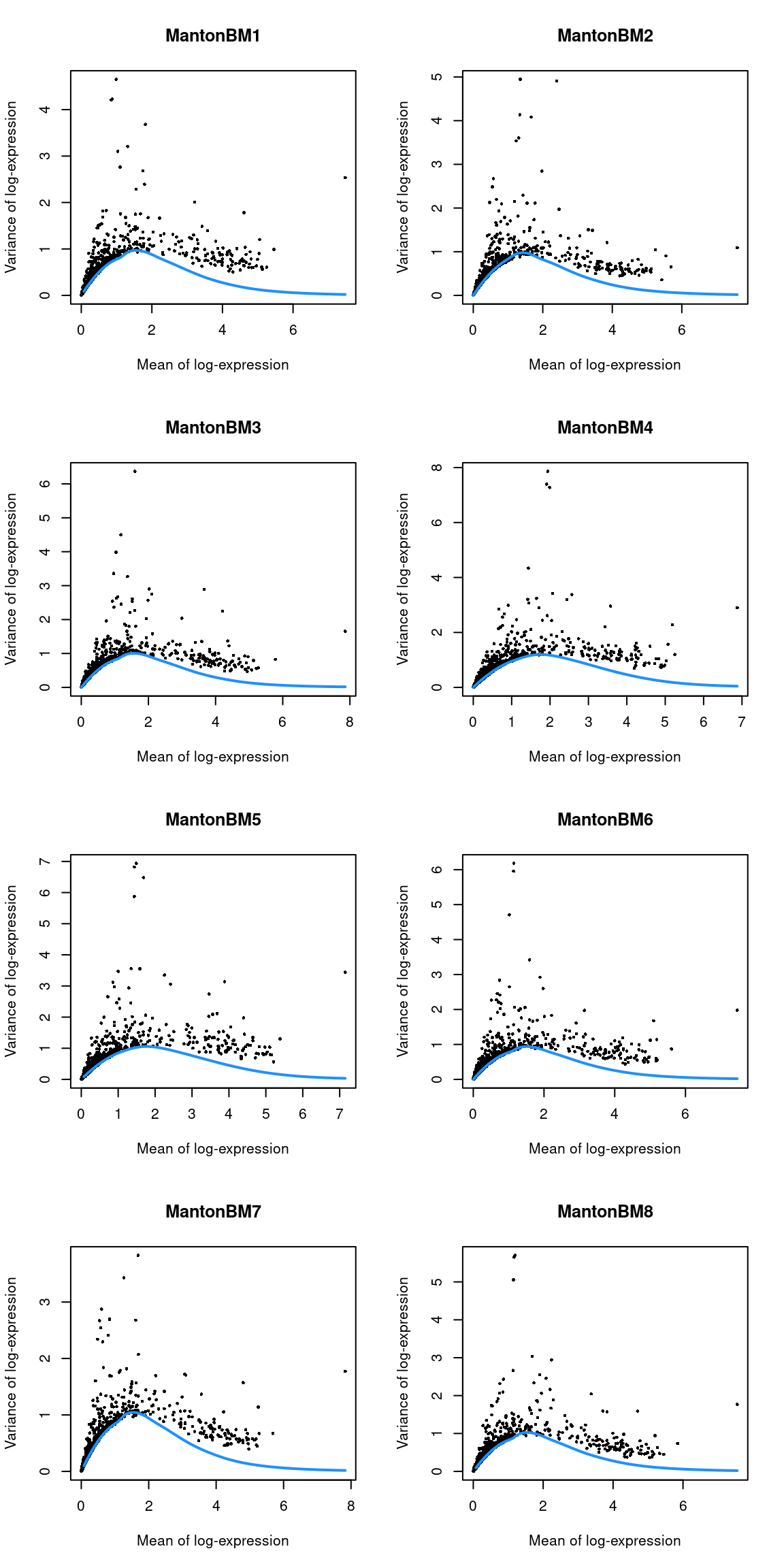
Figure 14.3: Per-gene variance as a function of the mean for the log-expression values in the HCA bone marrow dataset. Each point represents a gene (black) with the mean-variance trend (blue) fitted to the variances.
14.6 Data integration
Here we use multiple cores, randomized SVD and approximate nearest-neighbor detection to speed up this step.
library(batchelor)
library(BiocNeighbors)
set.seed(1010001)
merged.bone <- fastMNN(sce.bone, batch = sce.bone$Donor, subset.row = top.bone,
BSPARAM=BiocSingular::RandomParam(deferred = TRUE),
BNPARAM=AnnoyParam(),
BPPARAM=bpp)
reducedDim(sce.bone, 'MNN') <- reducedDim(merged.bone, 'corrected')We use the percentage of variance lost as a diagnostic measure:
## MantonBM1 MantonBM2 MantonBM3 MantonBM4 MantonBM5 MantonBM6 MantonBM7
## [1,] 0.006922 0.006392 0.000000 0.000000 0.000000 0.000000 0.000000
## [2,] 0.006380 0.006863 0.023049 0.000000 0.000000 0.000000 0.000000
## [3,] 0.005068 0.003084 0.005178 0.019496 0.000000 0.000000 0.000000
## [4,] 0.002009 0.001891 0.001901 0.001786 0.023105 0.000000 0.000000
## [5,] 0.002452 0.002003 0.001770 0.002926 0.002646 0.023852 0.000000
## [6,] 0.003167 0.003222 0.003169 0.002636 0.003362 0.003442 0.024650
## [7,] 0.001968 0.001701 0.002441 0.002045 0.001585 0.002312 0.002003
## MantonBM8
## [1,] 0.00000
## [2,] 0.00000
## [3,] 0.00000
## [4,] 0.00000
## [5,] 0.00000
## [6,] 0.00000
## [7,] 0.0321614.7 Dimensionality reduction
We set external_neighbors=TRUE to replace the internal nearest neighbor search in the UMAP implementation with our parallelized approximate search.
We also set the number of threads to be used in the UMAP iterations.
14.8 Clustering
Graph-based clustering generates an excessively large intermediate graph so we will instead use a two-step approach with \(k\)-means.
We generate 1000 small clusters that are subsequently aggregated into more interpretable groups with a graph-based method.
If more resolution is required, we can increase centers in addition to using a lower k during graph construction.
library(bluster)
set.seed(1000)
colLabels(sce.bone) <- clusterRows(reducedDim(sce.bone, "MNN"),
TwoStepParam(KmeansParam(centers=1000), NNGraphParam(k=5)))
table(colLabels(sce.bone))##
## 1 2 3 4 5 6 7 8 9 10 11 12 13
## 20331 11161 55464 47426 15731 10581 64721 26493 18703 15043 17097 4992 3157
## 14 15
## 3403 2422We observe mostly balanced contributions from different samples to each cluster (Figure 14.4), consistent with the expectation that all samples are replicates from different donors.
tab <- table(Cluster=colLabels(sce.bone), Donor=sce.bone$Donor)
library(pheatmap)
pheatmap(log10(tab+10), color=viridis::viridis(100))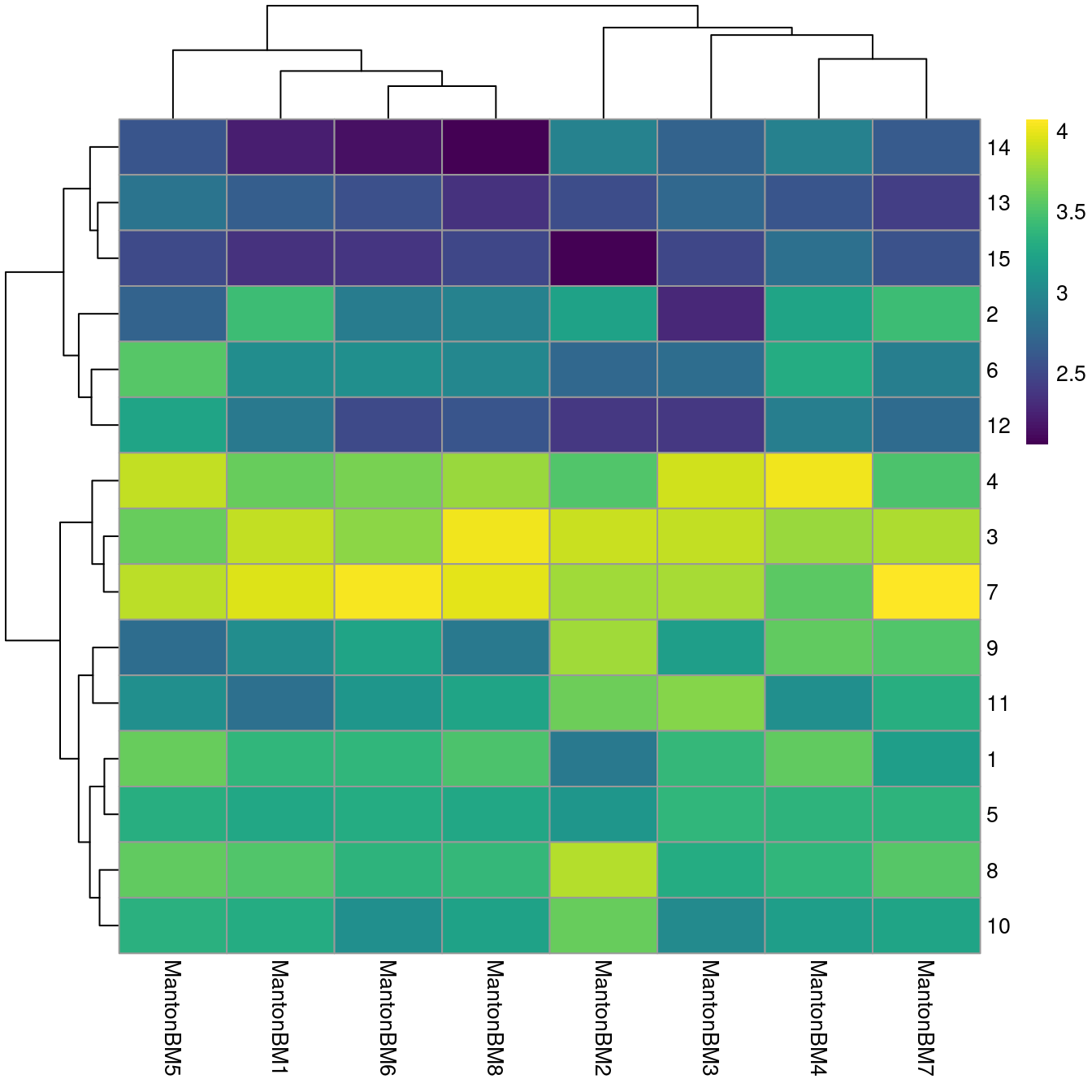
Figure 14.4: Heatmap of log10-number of cells in each cluster (row) from each sample (column).
# TODO: add scrambling option in scater's plotting functions.
scrambled <- sample(ncol(sce.bone))
gridExtra::grid.arrange(
plotUMAP(sce.bone, colour_by="label", text_by="label"),
plotUMAP(sce.bone[,scrambled], colour_by="Donor")
)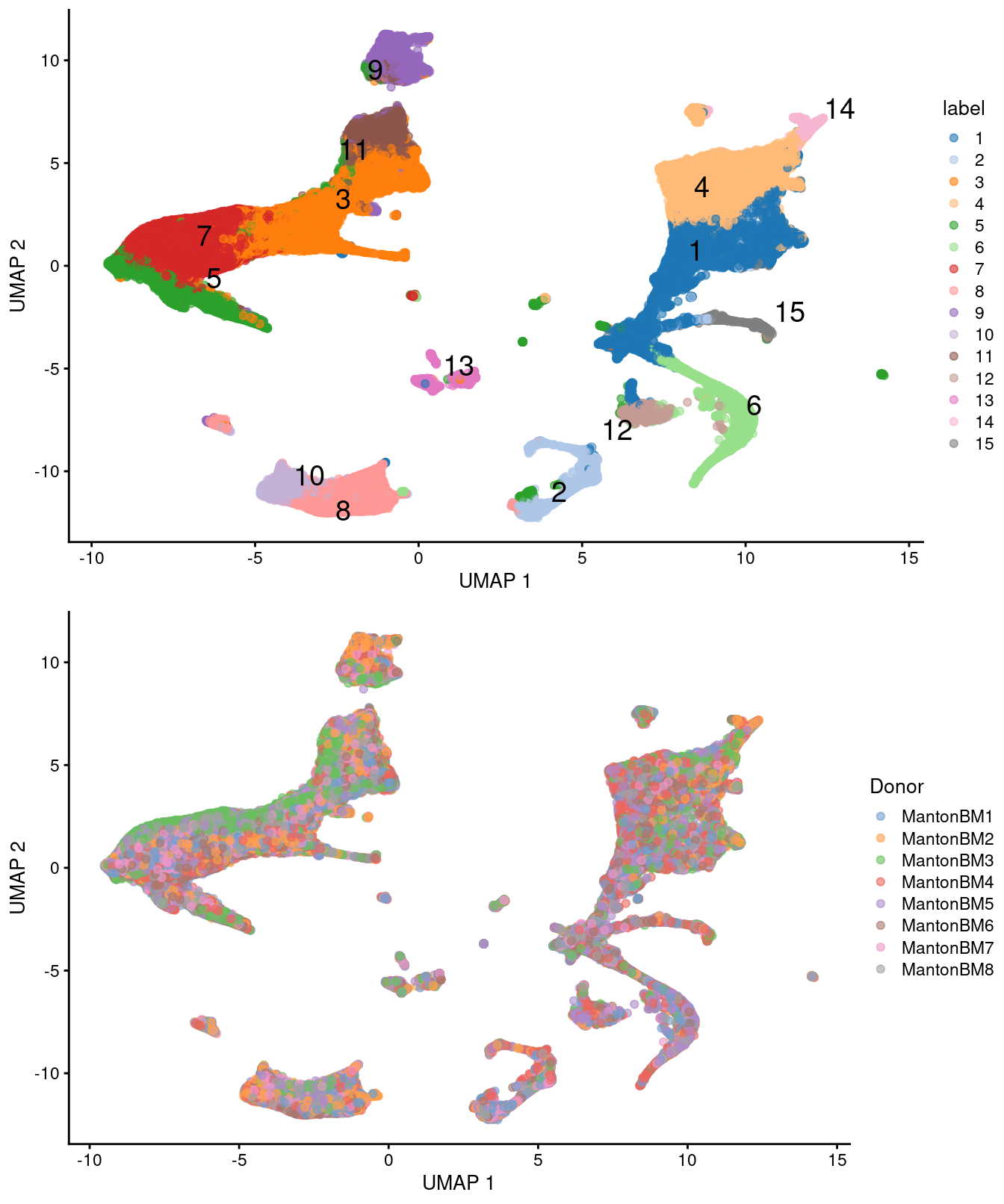
Figure 14.5: UMAP plots of the HCA bone marrow dataset after merging. Each point represents a cell and is colored according to the assigned cluster (top) or the donor of origin (bottom).
14.9 Differential expression
We identify marker genes for each cluster while blocking on the donor.
markers.bone <- findMarkers(sce.bone, block = sce.bone$Donor,
direction = 'up', lfc = 1, BPPARAM=bpp)We visualize the top markers for a randomly chosen cluster using a “dot plot” in Figure 14.6. The presence of upregulated genes like LYZ, S100A8 and VCAN is consistent with a monocyte identity for this cluster.
top.markers <- markers.bone[["4"]]
best <- top.markers[top.markers$Top <= 10,]
lfcs <- getMarkerEffects(best)
library(pheatmap)
pheatmap(lfcs, breaks=seq(-5, 5, length.out=101))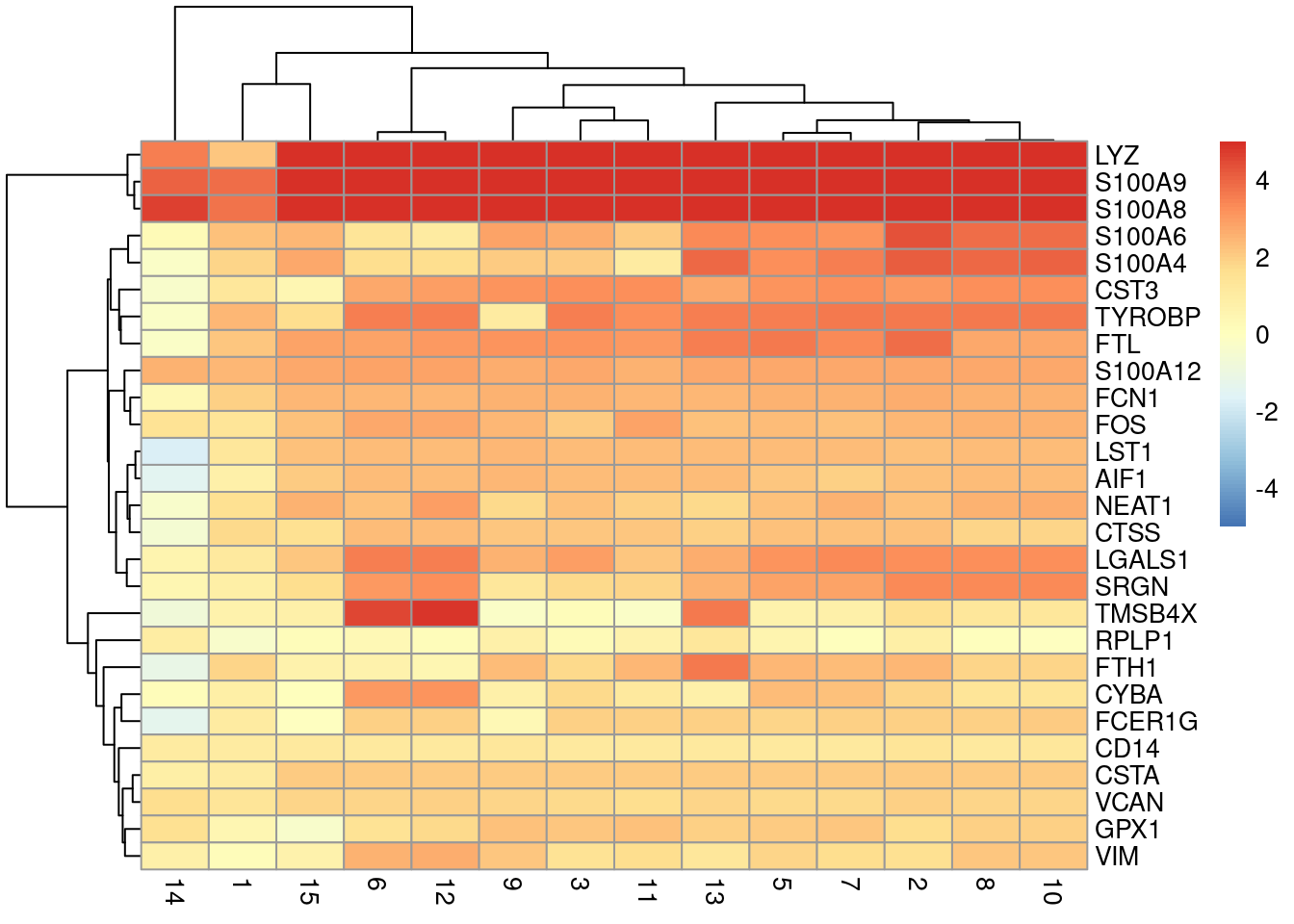
Figure 14.6: Heatmap of log2-fold changes for the top marker genes (rows) of cluster 4 compared to all other clusters (columns).
14.10 Cell type classification
We perform automated cell type classification using a reference dataset to annotate each cluster based on its pseudo-bulk profile. This is faster than the per-cell approaches described in Chapter 10.9 at the cost of the resolution required to detect heterogeneity inside a cluster. Nonetheless, it is often sufficient for a quick assignment of cluster identity, and indeed, cluster 4 is also identified as consisting of monocytes from this analysis.
se.aggregated <- sumCountsAcrossCells(sce.bone, id=colLabels(sce.bone), BPPARAM=bpp)
library(celldex)
hpc <- HumanPrimaryCellAtlasData()
library(SingleR)
anno.single <- SingleR(se.aggregated, ref = hpc, labels = hpc$label.main,
assay.type.test="sum")
anno.single## DataFrame with 15 rows and 5 columns
## scores first.labels tuning.scores
## <matrix> <character> <DataFrame>
## 1 0.366050:0.741975:0.637800:... GMP 0.676512:0.339887
## 2 0.399229:0.715314:0.628516:... Pro-B_cell_CD34+ 0.845406:0.776315
## 3 0.325812:0.654869:0.570039:... NK_cell 0.612492:0.483279
## 4 0.296455:0.742466:0.529404:... Pre-B_cell_CD34- 0.564310:0.254385
## 5 0.345773:0.565378:0.479722:... T_cells 0.616848:0.122525
## ... ... ... ...
## 11 0.326882:0.646229:0.561060:... NK_cell 0.546934:0.5190984
## 12 0.380710:0.684123:0.784540:... MEP 0.375942:0.3754427
## 13 0.368546:0.652935:0.580330:... B_cell 0.527988:0.3268898
## 14 0.294361:0.706019:0.527282:... Pre-B_cell_CD34- 0.600000:0.2447875
## 15 0.339786:0.687074:0.569933:... Pre-B_cell_CD34- 0.231710:0.0998799
## labels pruned.labels
## <character> <character>
## 1 GMP GMP
## 2 Pro-B_cell_CD34+ Pro-B_cell_CD34+
## 3 T_cells T_cells
## 4 Monocyte Monocyte
## 5 T_cells T_cells
## ... ... ...
## 11 T_cells T_cells
## 12 BM & Prog. BM & Prog.
## 13 B_cell NA
## 14 Monocyte Monocyte
## 15 GMP GMPSession Info
R version 4.2.1 (2022-06-23)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 20.04.5 LTS
Matrix products: default
BLAS: /home/biocbuild/bbs-3.15-bioc/R/lib/libRblas.so
LAPACK: /home/biocbuild/bbs-3.15-bioc/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_GB LC_COLLATE=C
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats4 stats graphics grDevices utils datasets methods
[8] base
other attached packages:
[1] SingleR_1.10.0 celldex_1.6.0
[3] pheatmap_1.0.12 bluster_1.6.0
[5] BiocNeighbors_1.14.0 batchelor_1.12.3
[7] scran_1.24.1 BiocParallel_1.30.4
[9] scater_1.24.0 ggplot2_3.3.6
[11] scuttle_1.6.3 EnsDb.Hsapiens.v86_2.99.0
[13] ensembldb_2.20.2 AnnotationFilter_1.20.0
[15] GenomicFeatures_1.48.4 AnnotationDbi_1.58.0
[17] rhdf5_2.40.0 HCAData_1.12.0
[19] SingleCellExperiment_1.18.1 SummarizedExperiment_1.26.1
[21] Biobase_2.56.0 GenomicRanges_1.48.0
[23] GenomeInfoDb_1.32.4 IRanges_2.30.1
[25] S4Vectors_0.34.0 BiocGenerics_0.42.0
[27] MatrixGenerics_1.8.1 matrixStats_0.62.0
[29] BiocStyle_2.24.0 rebook_1.6.0
loaded via a namespace (and not attached):
[1] AnnotationHub_3.4.0 BiocFileCache_2.4.0
[3] igraph_1.3.5 lazyeval_0.2.2
[5] digest_0.6.29 htmltools_0.5.3
[7] viridis_0.6.2 fansi_1.0.3
[9] magrittr_2.0.3 memoise_2.0.1
[11] ScaledMatrix_1.4.1 cluster_2.1.4
[13] limma_3.52.4 Biostrings_2.64.1
[15] prettyunits_1.1.1 colorspace_2.0-3
[17] blob_1.2.3 rappdirs_0.3.3
[19] ggrepel_0.9.1 xfun_0.33
[21] dplyr_1.0.10 crayon_1.5.2
[23] RCurl_1.98-1.9 jsonlite_1.8.2
[25] graph_1.74.0 glue_1.6.2
[27] gtable_0.3.1 zlibbioc_1.42.0
[29] XVector_0.36.0 DelayedArray_0.22.0
[31] BiocSingular_1.12.0 Rhdf5lib_1.18.2
[33] HDF5Array_1.24.2 scales_1.2.1
[35] edgeR_3.38.4 DBI_1.1.3
[37] Rcpp_1.0.9 viridisLite_0.4.1
[39] xtable_1.8-4 progress_1.2.2
[41] dqrng_0.3.0 bit_4.0.4
[43] rsvd_1.0.5 ResidualMatrix_1.6.1
[45] metapod_1.4.0 httr_1.4.4
[47] RColorBrewer_1.1-3 dir.expiry_1.4.0
[49] ellipsis_0.3.2 farver_2.1.1
[51] pkgconfig_2.0.3 XML_3.99-0.11
[53] uwot_0.1.14 CodeDepends_0.6.5
[55] sass_0.4.2 dbplyr_2.2.1
[57] locfit_1.5-9.6 utf8_1.2.2
[59] labeling_0.4.2 tidyselect_1.2.0
[61] rlang_1.0.6 later_1.3.0
[63] munsell_0.5.0 BiocVersion_3.15.2
[65] tools_4.2.1 cachem_1.0.6
[67] cli_3.4.1 generics_0.1.3
[69] RSQLite_2.2.18 ExperimentHub_2.4.0
[71] evaluate_0.17 stringr_1.4.1
[73] fastmap_1.1.0 yaml_2.3.5
[75] knitr_1.40 bit64_4.0.5
[77] purrr_0.3.5 KEGGREST_1.36.3
[79] sparseMatrixStats_1.8.0 mime_0.12
[81] xml2_1.3.3 biomaRt_2.52.0
[83] compiler_4.2.1 beeswarm_0.4.0
[85] filelock_1.0.2 curl_4.3.3
[87] png_0.1-7 interactiveDisplayBase_1.34.0
[89] statmod_1.4.37 tibble_3.1.8
[91] bslib_0.4.0 stringi_1.7.8
[93] highr_0.9 lattice_0.20-45
[95] ProtGenerics_1.28.0 Matrix_1.5-1
[97] vctrs_0.4.2 pillar_1.8.1
[99] lifecycle_1.0.3 rhdf5filters_1.8.0
[101] BiocManager_1.30.18 jquerylib_0.1.4
[103] cowplot_1.1.1 bitops_1.0-7
[105] irlba_2.3.5.1 httpuv_1.6.6
[107] rtracklayer_1.56.1 R6_2.5.1
[109] BiocIO_1.6.0 bookdown_0.29
[111] promises_1.2.0.1 gridExtra_2.3
[113] vipor_0.4.5 codetools_0.2-18
[115] assertthat_0.2.1 rjson_0.2.21
[117] withr_2.5.0 GenomicAlignments_1.32.1
[119] Rsamtools_2.12.0 GenomeInfoDbData_1.2.8
[121] parallel_4.2.1 hms_1.1.2
[123] grid_4.2.1 beachmat_2.12.0
[125] rmarkdown_2.17 DelayedMatrixStats_1.18.2
[127] shiny_1.7.2 ggbeeswarm_0.6.0
[129] restfulr_0.0.15 Bach, K., S. Pensa, M. Grzelak, J. Hadfield, D. J. Adams, J. C. Marioni, and W. T. Khaled. 2017. “Differentiation dynamics of mammary epithelial cells revealed by single-cell RNA sequencing.” Nat Commun 8 (1): 2128.
Grun, D., M. J. Muraro, J. C. Boisset, K. Wiebrands, A. Lyubimova, G. Dharmadhikari, M. van den Born, et al. 2016. “De Novo Prediction of Stem Cell Identity using Single-Cell Transcriptome Data.” Cell Stem Cell 19 (2): 266–77.
Lawlor, N., J. George, M. Bolisetty, R. Kursawe, L. Sun, V. Sivakamasundari, I. Kycia, P. Robson, and M. L. Stitzel. 2017. “Single-cell transcriptomes identify human islet cell signatures and reveal cell-type-specific expression changes in type 2 diabetes.” Genome Res. 27 (2): 208–22.
Lun, A. T. L., F. J. Calero-Nieto, L. Haim-Vilmovsky, B. Gottgens, and J. C. Marioni. 2017. “Assessing the reliability of spike-in normalization for analyses of single-cell RNA sequencing data.” Genome Res. 27 (11): 1795–1806.
Messmer, T., F. von Meyenn, A. Savino, F. Santos, H. Mohammed, A. T. L. Lun, J. C. Marioni, and W. Reik. 2019. “Transcriptional heterogeneity in naive and primed human pluripotent stem cells at single-cell resolution.” Cell Rep 26 (4): 815–24.
Muraro, M. J., G. Dharmadhikari, D. Grun, N. Groen, T. Dielen, E. Jansen, L. van Gurp, et al. 2016. “A Single-Cell Transcriptome Atlas of the Human Pancreas.” Cell Syst 3 (4): 385–94.
Nestorowa, S., F. K. Hamey, B. Pijuan Sala, E. Diamanti, M. Shepherd, E. Laurenti, N. K. Wilson, D. G. Kent, and B. Gottgens. 2016. “A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation.” Blood 128 (8): 20–31.
Paul, F., Y. Arkin, A. Giladi, D. A. Jaitin, E. Kenigsberg, H. Keren-Shaul, D. Winter, et al. 2015. “Transcriptional Heterogeneity and Lineage Commitment in Myeloid Progenitors.” Cell 163 (7): 1663–77.
Picelli, S., O. R. Faridani, A. K. Bjorklund, G. Winberg, S. Sagasser, and R. Sandberg. 2014. “Full-length RNA-seq from single cells using Smart-seq2.” Nat Protoc 9 (1): 171–81.
Pollen, A. A., T. J. Nowakowski, J. Shuga, X. Wang, A. A. Leyrat, J. H. Lui, N. Li, et al. 2014. “Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex.” Nat. Biotechnol. 32 (10): 1053–8.
Segerstolpe, A., A. Palasantza, P. Eliasson, E. M. Andersson, A. C. Andreasson, X. Sun, S. Picelli, et al. 2016. “Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes.” Cell Metab. 24 (4): 593–607.
Wajapeyee, N., S. Z. Wang, R. W. Serra, P. D. Solomon, A. Nagarajan, X. Zhu, and M. R. Green. 2010. “Senescence induction in human fibroblasts and hematopoietic progenitors by leukemogenic fusion proteins.” Blood 115 (24): 5057–60.
Zeisel, A., A. B. Munoz-Manchado, S. Codeluppi, P. Lonnerberg, G. La Manno, A. Jureus, S. Marques, et al. 2015. “Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq.” Science 347 (6226): 1138–42.
Zeng, H., E. H. Shen, J. G. Hohmann, S. W. Oh, A. Bernard, J. J. Royall, K. J. Glattfelder, et al. 2012. “Large-scale cellular-resolution gene profiling in human neocortex reveals species-specific molecular signatures.” Cell 149 (2): 483–96.
Zheng, G. X., J. M. Terry, P. Belgrader, P. Ryvkin, Z. W. Bent, R. Wilson, S. B. Ziraldo, et al. 2017. “Massively parallel digital transcriptional profiling of single cells.” Nat Commun 8 (January): 14049.